Description of functions in Chorus2¶
Major functions¶
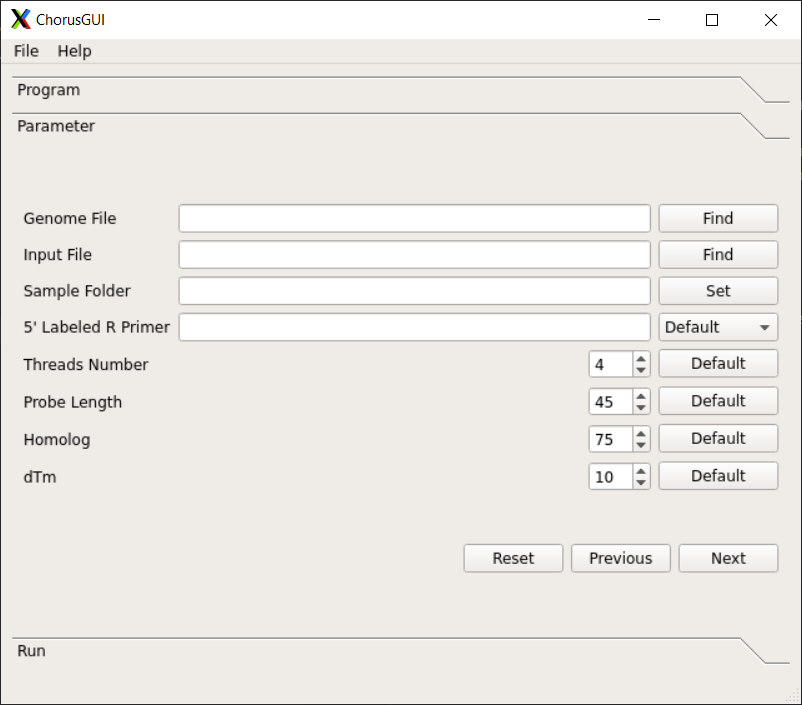
Chorus2¶
Function¶
Chorus2 Software for Oligo FISH probe design
Parameters¶
-j JELLYFISH, --jellyfish JELLYFISH
The path where Jellyfish software installed
-b BWA, --bwa BWA The path where BWA software installed
-g GENOME, --genome GENOME
Fasta format genome file, should include all sequences
from genome
-i INPUT, --input INPUT
Fasta format input file, can be whole genome, a
chromosome or one region from genome
-s SAVED, --save SAVED
The output folder for saving results
-p PRIMER, --primer PRIMER
A specific 5' labeled R primer for PCR reaction. For
example: CGTGGTCGCGTCTCA. (Default is none)
-t THREADS, --threads THREADS
Number of threads or CPUs to use. (Default: 1)
-l LENGTH, --length LENGTH
The probe length. (Default: 45)
--homology HOMOLOGY The maximum homology(%) between target sequence and
probe, range from 50 to 100. (Default: 75)
-d DTM, --dtm DTM The minimum value of dTm (hybrid Tm - hairpin Tm),
range from 0 to 37. (Default: 10)
--skipdtm SKIPDTM skip calculate dtm, for oligo longer than 50.
--step STEP The step length for k-mer searching in a sliding
window, step length>=1. (Default: 5)
--docker DOCKER Only used in Docker version of Chorus
--ploidy PLOIDY The ploidy of the given genome (test version).
(Default: 2)
Usage¶
$ Chorus2 -i TAIR10_chr_all.fas -g TAIR10_chr_all.fas -t 4 \
-j /opt/software/jellyfish/bin/jellyfish -b /opt/software/bwa/bwa -s sample
ChorusNGSfilter¶
Function¶
ChorusNGSfilter for counting Oligo FISH probe k-mer score using NGS data
Parameters¶
-j JELLYFISH, --jellyfish JELLYFISH
The path where Jellyfish software installed
-g GENOME, --genome GENOME
Fasta format genome file, should include all sequences
from genome
-i INPUT, --input INPUT
Fastq format input files contain reads from whole
genome shotgun sequencing, files can be gzipped.
Multiple files separate with ",". For example:
1.fq.gz,2.fq.gz
-jfile JFILE, --jellyfishfile JFILE
prebuild jellyfish index file, conflict with input
argument.
-z {gz,text}, --gzipped {gz,text}
Input fastq file is gzipped(gz) or uncompressed(text).
(Default: gz)
-t THREADS, --threads THREADS
Number of threads or CPUs to use. (Default: 1)
-k KMER, --kmer KMER Length of k-mer used for counting k-mers in input
fastq files. (Default: 17)
-p PROBE, --probe PROBE
The bed format probe file generated by Chorus
-o OUTPUT, --output OUTPUT
Output bed format probe file with k-mer score.
(Default: output.bed)
Usage¶
$ ChorusNGSfilter -i 1.fq.gz,2.fq.gz -z gz -t 4 -g TAIR10_chr_all.fas \
-j /opt/software/jellyfish/bin/jellyfish -p probe.bed -o output.bed
ChorusNGSselect¶
Function¶
ChorusNGSselect for Oligo FISH probe selection by NGS k-mer score
Parameters¶
-i INPUT, --input INPUT
Input bed format probe file generated by
ChorusNGSfilter
-o OUTPUT, --output OUTPUT
Output bed format probe file after k-mer score filter.
(Default: filtered_output.bed)
-m MINK, --min MINK Minimum k-mer score, score < min value will be
dropped. For example: 900. Incompatible with parameter
'-q/-p' (Default: 0)
-l MAXK, --max MAXK Maximum k-mer score, score > max value will be
dropped. For example: 2000. Incompatible with
parameter '-q/-p' (Default: 10000000)
-q MINQUANTILE, --minquantile MINQUANTILE
Filter < min% quantile k-mer score, range from 0 to 1.
For example: 0.25 means 25% quantile. Incompatible
with parameter '-m/-l'. (Default: 0.1)
-p MAXQUANTILE, --maxquantile MAXQUANTILE
Filter > max% quantile k-mer score, range from 0 to 1.
For example: 0.75 means 75% quantile. Incompatible
with parameter '-m/-l'. (Default: 0.9)
-bs, --bothstrand Keep both + and - strand probes. (Default is True)
-ss, --singlestrand Keep only + strand probes. Incompatible with parameter
'-bs/--bothstrand'
-d DIS, --distance DIS
Minimum distance between two adjacent probes.
(Default: 25)
Usage¶
$ ChorusNGSselect -i ChorusNGSfilter_output.bed -q 0.1 -p 0.9 -d 25 \
-o filtered_output.bed
ChorusHomo¶
Function¶
ChorusHomo for finding probes which can hybridize to a close related species
Parameters¶
-j JELLYFISH, --jellyfish JELLYFISH
The path where Jellyfish software installed
-b BWA, --bwa BWA The path where BWA software installed
-ga SOURCE, --source SOURCE
Fasta format genome file (GenomeA) which the probe
were generated from, should include all sequences from
genome
-gb TARGET, --target TARGET
Fasta format genome file (GenomeB) which the probe
will be aligned to, should include all sequences from
genome
-i INPUT, --input INPUT
BED format input file, contains oligo probes generated
from Chorus2
-s SAVED, --save SAVED
The output folder for saving results
-t THREADS, --threads THREADS
Number of threads or CPUs to use. (Default: 1)
Usage¶
$ ChorusHomo -i probe.bed -ga source_genome.fasta -gb target_genome.fasta \
-j /opt/software/jellyfish/bin/jellyfish -b /opt/software/bwa/bwa \
-t 4 -s sample
ChorusNoRef¶
Function¶
ChorusNoRef for designing oligo-FISH probe for no reference genome
Parameters¶
-j JELLYFISH, --jellyfish JELLYFISH
The path where Jellyfish software installed
-b BWA, --bwa BWA The path where BWA software installed
-c BCFTOOLS, --bcftools BCFTOOLS
The path where bcftools software installed
-m SAMTOOLS, --samtools SAMTOOLS
The path where samtools software installed
-g GENOME, --genome GENOME
Fasta format genome file, should include all sequences
from genome
-s SAVED, --save SAVED
The output folder for saving results
--tmp TMP The temporary fold for processing
-p PROBE, --probe PROBE
Original probe design by Chorus2 and filtered by
ChorusNGSfilter
-r1 READS1, --reads1 READS1
read1 of species, example: For one Species only:
species_R1.fq; for more than one species:
species1_R1.fq,species2_R1.fq
-r2 READS2, --reads2 READS2
read1 of species, example: For one Species only:
species_R2.fq; for more than one species:
species1_R2.fq,species2_R2.fq
-n NAMES, --names NAMES
species name(s), the order must same as r1, r2
-t THREADS, --threads THREADS
Number of threads or CPUs to use. (Default: 1)
--minkmer MINKMER Probe min count for illumina reads
-l LENGTH, --length LENGTH
The probe length. (Default: 45)
-d MINDEPTH, --mindepth MINDEPTH
Minimum depth covered by illumina sequences. (Default: 3)
Usage¶
$ ChorusNoRef -g related_genome.fasta -s results -p probes.bed \
-r1 species1_R1.fq,species2_R1.fq -r2 species1_R2.fq,species2_R2.fq \
-n species1,species2 -t 4 --minkmer 0 -l 45 -d 3
Miscellaneous functions¶
ChorusDraftPrebuild¶
Function¶
ChorusDraftPrebuild for combining short sequence to speed up oligo search
Parameters¶
-i INPUT, --input INPUT
Fasta format input file contains short sequences
-o OUTPUT, --output OUTPUT
Fasta format output file with combined long sequences
for speeding up oligo search. (default: output.fa)
Usage¶
$ ChorusDraftPrebuild -i short_sequences.fasta -o long_sequences.fasta