Install¶
Docker (For Unix & Windows)¶
Install Docker
Terminal version¶
Download Chorus2:
$ docker pull forrestzhang/docker-chorus
$ docker run -v $PWD:/home/chorus -e CHORUS_USER=$USER -e CHORUS_UID=$UID forrestzhang/docker-chorus -h
usage: Chorus [-h] [--version] [-j JELLYFISH] [-b BWA] -g GENOME -i INPUT
[-s SAVED] [-p PRIMER] [-t THREADS] [-l LENGTH]
[--homology HOMOLOGY] [-d DTM] [--docker DOCKER]
Chorus Software for Oligo FISH probe design
optional arguments:
-h, --help show this help message and exit
--version show program's version number and exit
-j JELLYFISH, --jellyfish JELLYFISH
jellyfish path
-b BWA, --bwa BWA bwa path
-g GENOME, --genome GENOME
fasta format genome file
-i INPUT, --input INPUT
fasta format input file
-s SAVED, --save SAVED
result saved folder
-p PRIMER, --primer PRIMER
5' labeled R primer
-t THREADS, --threads THREADS
threads number or how may cpu you wanna use
-l LENGTH, --length LENGTH
probe length
--homology HOMOLOGY homology, from 50 to 100
-d DTM, --dtm DTM dTm, from 0 to 37
--docker DOCKER
GUI version¶
Download Chorus2:
$ docker pull forrestzhang/chorus-gui
$ docker run -i -t -p 6080:80 -v $PWD:/root/Desktop/Data forrestzhang/chorus-gui
Open your web browse, enter http://localhost:6080
In your web browse, open a LXTerminal
$ python3 /opt/software/Chorus/ChorusGUI.py
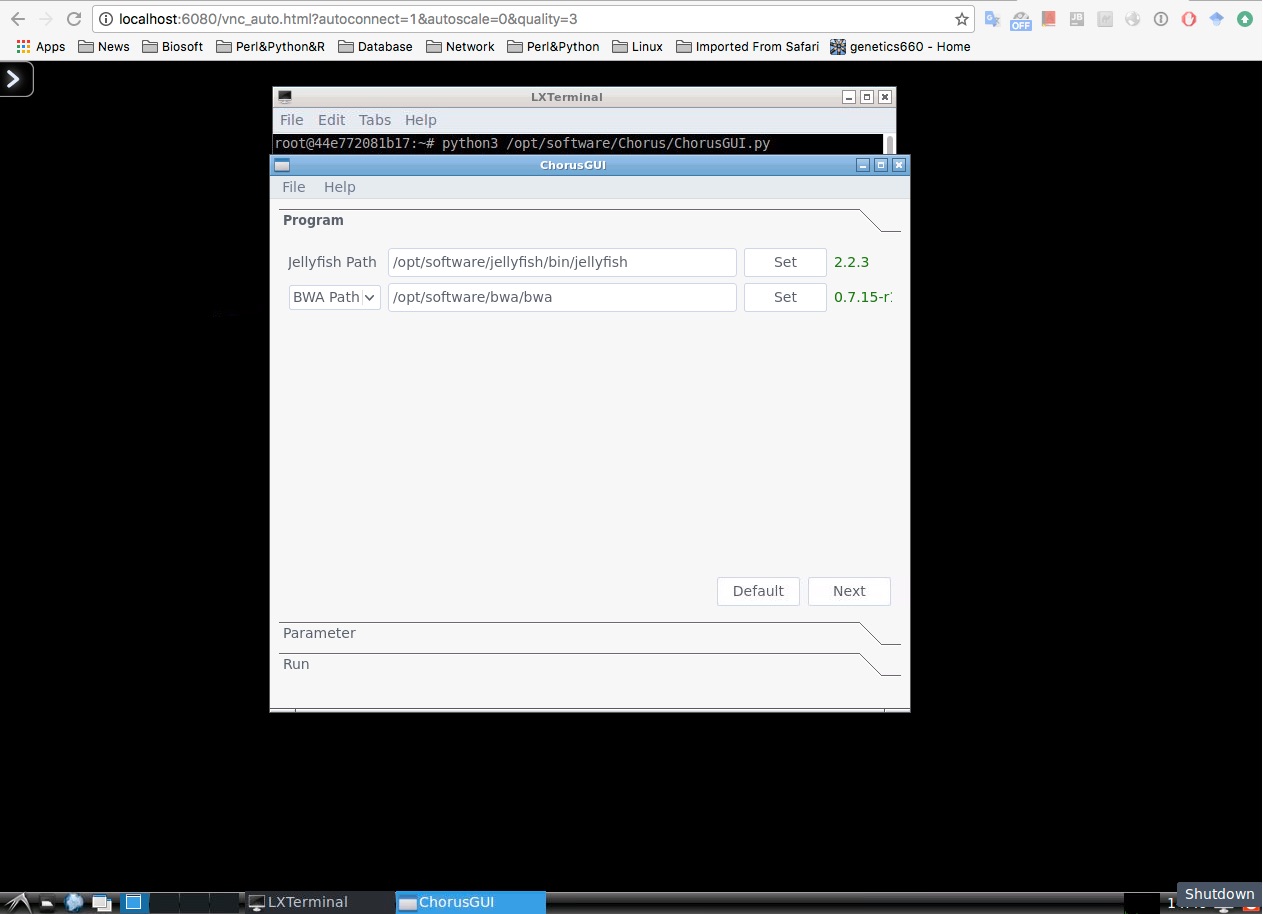
Manually Install¶
Linux (Ubuntu 18.04)¶
Manually install all packages¶
Install dependent package
$ apt-get update && apt-get install -y build-essential \
cython3 \
zlib1g-dev \
zlibc \
openjdk-7-jre \
git \
libboost-dev \
autoconf \
libncursesw5-dev \
libncurses5 \
ncurses-dev \
libboost-thread-dev \
python3-pip \
samtools \
bcftools \
unzip \
python \
curl \
wget
Install jellyfish
$ mkdir /opt/software
$ cd /opt/software
$ wget https://github.com/gmarcais/Jellyfish/releases/download/v2.3.0/jellyfish-2.3.0.tar.gz
$ tar zxvf jellyfish-2.3.0.tar.gz
$ mv jellyfish-2.3.0 jellyfish
$ cd jellyfish
$ ./configure && make && make install
Install bwa
$ cd /opt/software
$ git clone https://github.com/lh3/bwa.git
$ cd bwa
$ make
Install primer3-py
$ cd /opt/software
$ wget https://github.com/forrestzhang/primer3-py/archive/unicode.zip
$ unzip unicode.zip
$ cd primer3-py-unicode
$ python3 setup.py install
Download Chorus2 and test the terminal version
$ cd /opt/software
$ git clone https://github.com/zhangtaolab/Chorus2.git
$ pip install -r /opt/software/Chorus2/requirements.txt
$ python3 /opt/software/Chorus2/Chorus2.py -h
usage: Chorus2 [-h] [--version] [-j JELLYFISH] [-b BWA] -g GENOME -i INPUT
[-s SAVED] [-p PRIMER] [-t THREADS] [-l LENGTH]
[--homology HOMOLOGY] [-d DTM] [--skipdtm SKIPDTM]
[--step STEP] [--docker DOCKER] [--ploidy PLOIDY]
Chorus2 Software for Oligo FISH probe design
optional arguments:
-h, --help show this help message and exit
--version show program's version number and exit
-j JELLYFISH, --jellyfish JELLYFISH
The path where Jellyfish software installed
-b BWA, --bwa BWA The path where BWA software installed
-g GENOME, --genome GENOME
Fasta format genome file, should include all sequences
from genome
-i INPUT, --input INPUT
Fasta format input file, can be whole genome, a
chromosome or one region from genome
-s SAVED, --save SAVED
The output folder for saving results
-p PRIMER, --primer PRIMER
A specific 5' labeled R primer for PCR reaction. For
example: CGTGGTCGCGTCTCA. (Default is none)
-t THREADS, --threads THREADS
Number of threads or CPUs to use. (Default: 1)
-l LENGTH, --length LENGTH
The probe length. (Default: 45)
--homology HOMOLOGY The maximum homology(%) between target sequence and
probe, range from 50 to 100. (Default: 75)
-d DTM, --dtm DTM The minimum value of dTm (hybrid Tm - hairpin Tm),
range from 0 to 37. (Default: 10)
--skipdtm SKIPDTM skip calculate dtm, for oligo longer than 50.
--step STEP The step length for k-mer searching in a sliding
window, step length>=1. (Default: 5)
--docker DOCKER Only used in Docker version of Chorus
--ploidy PLOIDY The ploidy of the given genome (test version).
(Default: 2)
Example:
Chorus2 -i TAIR10_chr_all.fas -g TAIR10_chr_all.fas -t 4 \
-j /opt/software/jellyfish/bin/jellyfish -b /opt/software/bwa/bwa -s sample
Install GUI dependencies and test the GUI version
$ cd /opt/software/
$ pip install -r /opt/software/Chorus2/requirements_GUI.txt
$ python3 /opt/software/Chorus2/ChorusGUI.py
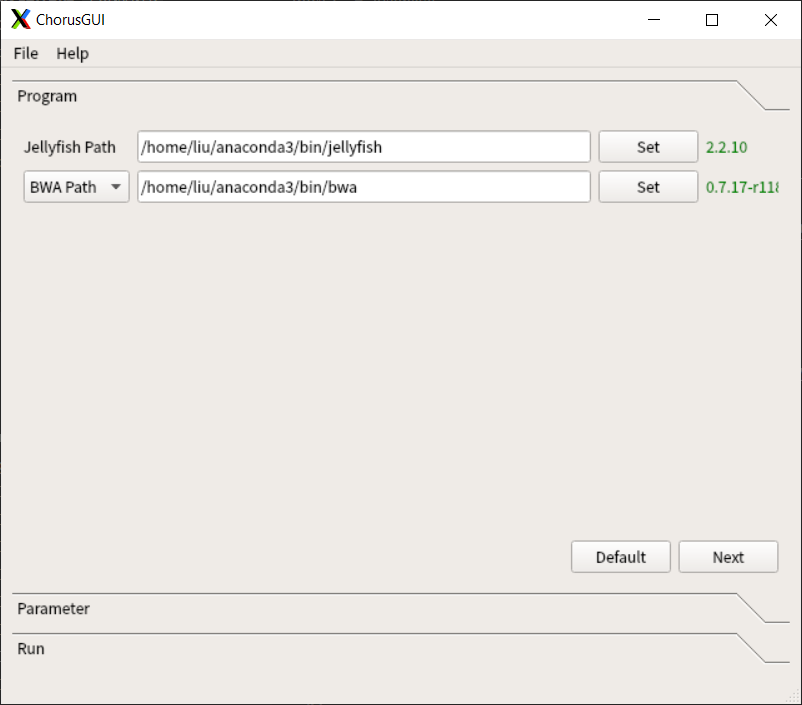
Install Chorus2 directly by Anaconda (Recommended)¶
Download and Install Anaconda (python 3.x verison for linux)
$ cd ~
$ wget https://repo.anaconda.com/archive/Anaconda3-xxxx-Linux-x86_64.sh
$ sh Anaconda3-xxxx-Linux-x86_64.sh
Add bioconda channel
$ conda config --add channels conda-forge
$ conda config --add channels defaults
$ conda config --add channels bioconda
Install Chorus2 by conda
$ conda install chorus2
$ Chorus2 -h
usage: Chorus2 [-h] [--version] [-j JELLYFISH] [-b BWA] -g GENOME -i INPUT
[-s SAVED] [-p PRIMER] [-t THREADS] [-l LENGTH]
[--homology HOMOLOGY] [-d DTM] [--skipdtm SKIPDTM]
[--step STEP] [--docker DOCKER] [--ploidy PLOIDY]
Chorus2 Software for Oligo FISH probe design
optional arguments:
-h, --help show this help message and exit
--version show program's version number and exit
-j JELLYFISH, --jellyfish JELLYFISH
The path where Jellyfish software installed
-b BWA, --bwa BWA The path where BWA software installed
-g GENOME, --genome GENOME
Fasta format genome file, should include all sequences
from genome
-i INPUT, --input INPUT
Fasta format input file, can be whole genome, a
chromosome or one region from genome
-s SAVED, --save SAVED
The output folder for saving results
-p PRIMER, --primer PRIMER
A specific 5' labeled R primer for PCR reaction. For
example: CGTGGTCGCGTCTCA. (Default is none)
-t THREADS, --threads THREADS
Number of threads or CPUs to use. (Default: 1)
-l LENGTH, --length LENGTH
The probe length. (Default: 45)
--homology HOMOLOGY The maximum homology(%) between target sequence and
probe, range from 50 to 100. (Default: 75)
-d DTM, --dtm DTM The minimum value of dTm (hybrid Tm - hairpin Tm),
range from 0 to 37. (Default: 10)
--skipdtm SKIPDTM skip calculate dtm, for oligo longer than 50.
--step STEP The step length for k-mer searching in a sliding
window, step length>=1. (Default: 5)
--docker DOCKER Only used in Docker version of Chorus
--ploidy PLOIDY The ploidy of the given genome (test version).
(Default: 2)
Example:
Chorus2 -i TAIR10_chr_all.fas -g TAIR10_chr_all.fas -t 4 \
-j /opt/software/jellyfish/bin/jellyfish -b /opt/software/bwa/bwa -s sample
Test the GUI version
$ ChorusGUI
MacOS¶
Install Chorus2 directly by Anaconda (Recommended)¶
Download and Install Anaconda (python 3.x Command Line Installer for MacOS)
$ cd ~
$ wget https://repo.anaconda.com/archive/Anaconda3-xxxx-MacOSX-x86_64.sh
$ sh Anaconda3-xxxx-MacOSX-x86_64.sh
Add bioconda channel
$ conda config --add channels conda-forge
$ conda config --add channels defaults
$ conda config --add channels bioconda
Install Chorus2 by conda
$ conda install chorus2
$ Chorus2 -h
usage: Chorus2 [-h] [--version] [-j JELLYFISH] [-b BWA] -g GENOME -i INPUT
[-s SAVED] [-p PRIMER] [-t THREADS] [-l LENGTH]
[--homology HOMOLOGY] [-d DTM] [--skipdtm SKIPDTM]
[--step STEP] [--docker DOCKER] [--ploidy PLOIDY]
Chorus2 Software for Oligo FISH probe design
optional arguments:
-h, --help show this help message and exit
--version show program's version number and exit
-j JELLYFISH, --jellyfish JELLYFISH
The path where Jellyfish software installed
-b BWA, --bwa BWA The path where BWA software installed
-g GENOME, --genome GENOME
Fasta format genome file, should include all sequences
from genome
-i INPUT, --input INPUT
Fasta format input file, can be whole genome, a
chromosome or one region from genome
-s SAVED, --save SAVED
The output folder for saving results
-p PRIMER, --primer PRIMER
A specific 5' labeled R primer for PCR reaction. For
example: CGTGGTCGCGTCTCA. (Default is none)
-t THREADS, --threads THREADS
Number of threads or CPUs to use. (Default: 1)
-l LENGTH, --length LENGTH
The probe length. (Default: 45)
--homology HOMOLOGY The maximum homology(%) between target sequence and
probe, range from 50 to 100. (Default: 75)
-d DTM, --dtm DTM The minimum value of dTm (hybrid Tm - hairpin Tm),
range from 0 to 37. (Default: 10)
--skipdtm SKIPDTM skip calculate dtm, for oligo longer than 50.
--step STEP The step length for k-mer searching in a sliding
window, step length>=1. (Default: 5)
--docker DOCKER Only used in Docker version of Chorus
--ploidy PLOIDY The ploidy of the given genome (test version).
(Default: 2)
Example:
Chorus2 -i TAIR10_chr_all.fas -g TAIR10_chr_all.fas -t 4 \
-j /opt/software/jellyfish/bin/jellyfish -b /opt/software/bwa/bwa -s sample
Test the GUI version
$ ChorusGUI
Windows 10 (WSL)¶
Install WSL (Windows Subsystem for Linux)¶
Open WSL in Control Panel - Programs and Features - Turn Windows features on or off
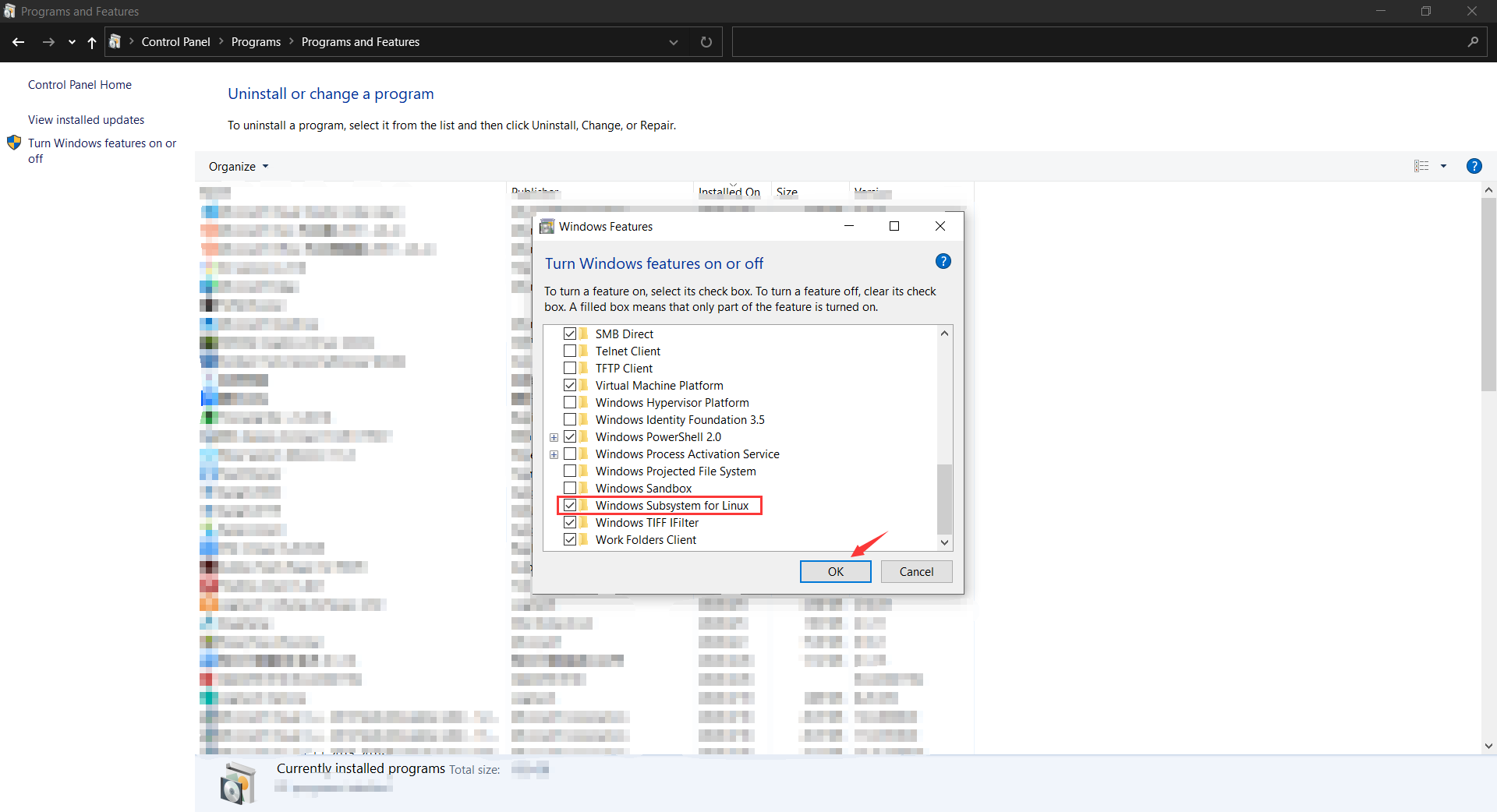
After reboot computer, install Ubuntu 18.04 LTS in Microsoft Store

Launch Ubuntu 18.04 LTS App, Initiate the WSL (Ubuntu 18.04)
$ sudo apt update
$ sudo apt upgrage
$ sudo apt install wget
Install Chorus2 directly by Anaconda¶
Download and Install Anaconda (python 3.x verison for linux)
$ cd ~
$ wget https://repo.anaconda.com/archive/Anaconda3-xxxx-Linux-x86_64.sh
$ sh Anaconda3-xxxx-Linux-x86_64.sh
Add bioconda channel
$ conda config --add channels conda-forge
$ conda config --add channels defaults
$ conda config --add channels bioconda
Install Chorus2 by conda
$ conda install chorus2
$ Chorus2 -h
usage: Chorus2 [-h] [--version] [-j JELLYFISH] [-b BWA] -g GENOME -i INPUT
[-s SAVED] [-p PRIMER] [-t THREADS] [-l LENGTH]
[--homology HOMOLOGY] [-d DTM] [--skipdtm SKIPDTM]
[--step STEP] [--docker DOCKER] [--ploidy PLOIDY]
Chorus2 Software for Oligo FISH probe design
optional arguments:
-h, --help show this help message and exit
--version show program's version number and exit
-j JELLYFISH, --jellyfish JELLYFISH
The path where Jellyfish software installed
-b BWA, --bwa BWA The path where BWA software installed
-g GENOME, --genome GENOME
Fasta format genome file, should include all sequences
from genome
-i INPUT, --input INPUT
Fasta format input file, can be whole genome, a
chromosome or one region from genome
-s SAVED, --save SAVED
The output folder for saving results
-p PRIMER, --primer PRIMER
A specific 5' labeled R primer for PCR reaction. For
example: CGTGGTCGCGTCTCA. (Default is none)
-t THREADS, --threads THREADS
Number of threads or CPUs to use. (Default: 1)
-l LENGTH, --length LENGTH
The probe length. (Default: 45)
--homology HOMOLOGY The maximum homology(%) between target sequence and
probe, range from 50 to 100. (Default: 75)
-d DTM, --dtm DTM The minimum value of dTm (hybrid Tm - hairpin Tm),
range from 0 to 37. (Default: 10)
--skipdtm SKIPDTM skip calculate dtm, for oligo longer than 50.
--step STEP The step length for k-mer searching in a sliding
window, step length>=1. (Default: 5)
--docker DOCKER Only used in Docker version of Chorus
--ploidy PLOIDY The ploidy of the given genome (test version).
(Default: 2)
Example:
Chorus2 -i TAIR10_chr_all.fas -g TAIR10_chr_all.fas -t 4 \
-j /opt/software/jellyfish/bin/jellyfish -b /opt/software/bwa/bwa -s sample